近日,中国科学技术大学李微雪教授结合物理启发的可解释机器学习算法与第一性原理计算,解决了一个多相催化研究中长期存在的关于催化结构敏感性难题。研究成果于近日以“Structure Sensitivity of Metal Catalysts Revealed by Interpretable Machine Learning and First-principles Calculations”为题发表于《美国化学会》期刊(J. Am. Chem. Soc.)。
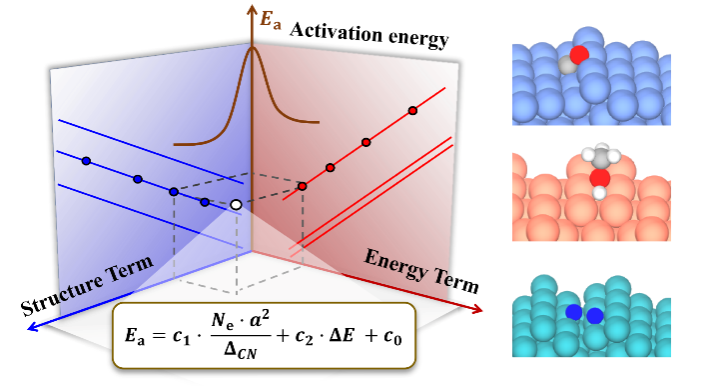
催化反应活性位及其结构敏感性是多相催化研究中最为重要的基本概念之一。尽管近年来研究取得了很大进展,但由于影响因素众多并横跨多个空间和时间尺度,如何在原子尺度上确定催化反应的活性位及其结构敏感性,依然是催化材料理性设计中所面临的一大挑战。举例来说,活化能与反应热之间的Brønsted–Evans–Polanyi(BEP)关系以及不同分子吸附能之间的线性标度律,长期以来被视为催化反应机理和催化优化设计的最重要的基本研究框架。但是,由于BEP关系和标度律中缺乏催化剂几何结构和化学组分的明确信息,这使它们原则上无法描述催化结构敏感性,从而极大限制了催化剂的优化设计研究。
机器学习方法在多相催化研究中发挥着日益重要的作用,并被应用到催化剂的结构敏感性研究中。但迄今为止大多数研究都属于端到端的“黑盒子”研究,研究结果缺乏很好的物理可解释性。物理上具有清晰的可解释性、明确包含催化剂的几何结构和化学组分、并能准确预测催化反应能垒的解析关系,目前仍然亟待建立。另外,由于催化反应能垒的计算主要通过高精度、高成本的密度泛函理论来完成,系统的理论数据也较为匮乏。因此,经常需要参考不同的数据源,数据源的多样性所带来的挑战也需要采取合适的机器学习算法。
面对这一问题,在本研究工作中,作者基于物理启发的可解释多任务学习符号回归和包含多样性的第一性原理计算数据集,在领域知识和化学直觉的基础上,建立了一个简洁、物理图像清晰的描述符。该描述符由催化剂的结构项和催化反应的能量项两部分组成,可用于准确预测各种分子在不同组分和结构金属催化剂上的活化能垒。其中,新建立的结构项由催化剂的拓扑配位不饱和度、价电子和晶格常数三个变量组成,借此成功破解了金属催化剂的结构敏感性问题,并突显了数据驱动理论模型的透明度(“白盒子”研究)在构建催化物理模型方面的重要性。
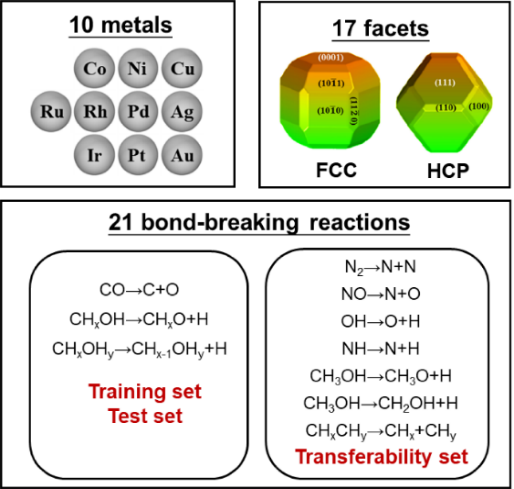
图1 涵盖了多种金属、晶面、晶相和反应的第一性原理计算数据集
在具体机器学习建模过程中,该工作提出了几点关键策略和思路。首先,为保障数据集的多样性,构建了一套涵盖了21种不同化学键断键能垒、10种过渡金属催化剂、2种不同晶相和17种不同晶面的较大多源数据集(图1)。其次,根据领域知识和化学直觉,推断表面能与活化能应该存在很强关联,而表面能又与表面悬挂键或配位不饱和度存在关联。在综合考虑表面暴露各种不同配位原子贡献的情况下,作者定义了一个新的、纯粹反映催化剂结构特征的物理量:拓扑配位不饱和度ΔCN
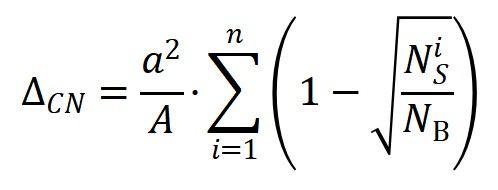
图2a显示,催化反应的活化能与拓扑配位不饱和度的相关性明显优于表面能和传统BEP中的反应热。将这一拓扑配位不饱和度,与反应热以及催化剂的其它基本原子参数作为物理特征一起放到相应的机器学习研究中。第三,为保障数据驱动模型的可解释性、准确性和通用性等,考虑到多源数据可能含有的非自洽性,以及不同分子之间的差异难以显式描述等情景,该工作采用了多任务学习符号回归策略进行机器学习建模。
机器学习结果最终给出的分子活化能最佳模型如下:
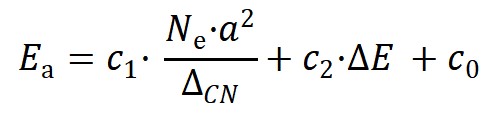
其中Ne为金属催化剂的价电子数,a为相应的晶格常数,ΔE为反应热,c1,c2和c0是对应的系数。在该二维模型中,第一项为结构项,正比于价电子数和晶格常数平方大小,反比于拓扑配位不饱和度,第二项为经典BEP关系中的反应能项。该模型能够准确预测分子断键能垒,并在具有不同对称性、键级和空间位阻等化学键的数据集上表现出良好的普适性(图2b和2c)。
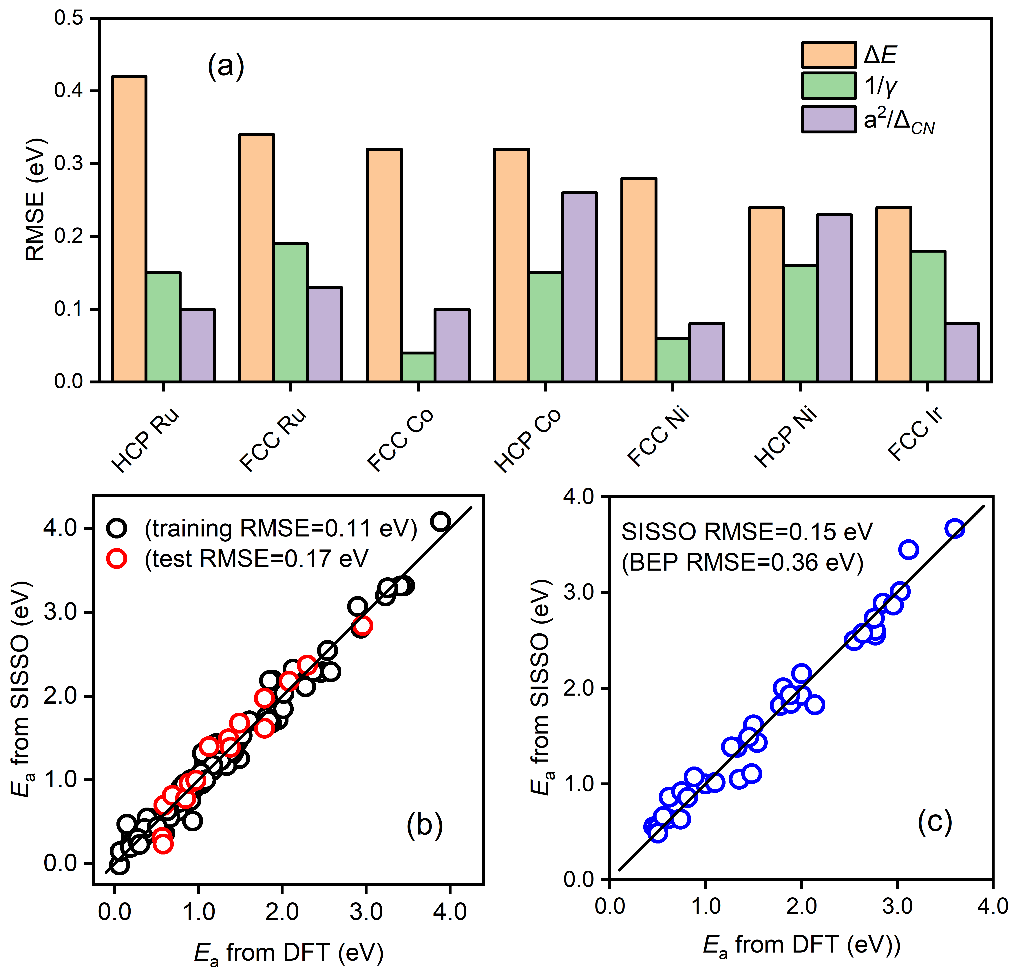
图2 (a)CO活化能垒与不同物理参数(反应热、表面能、拓扑配位不饱和度)之间的统计相关性;(b)机器学习模型的训练和测试准确度;(c)模型在新体系上的迁移性预测能力
上述方程由于明确包含了催化剂的组分、结构和反应热信息,因此活化能对组分调制的结构项(图3a)和能量项(图3b)的依赖关系可用来拆分催化剂的几何效应和电子效应。同时活化能对这两项投影的大小可用来对催化结构敏感性进行分类。如图3c所示,活化能在前者较大的投影和系数意味着该分子(比如CO, NO, N2)的活化过程是一种结构敏感的反应,而如果在后者上(比如OH,NH)则意味着该反应为结构不敏感,这一结论主要是适合于小分子。较大的分子因其空间位阻效应显著,相应的投影不能用来判断反应的结构敏感性,但相应公式的预测能力依然表现出色。
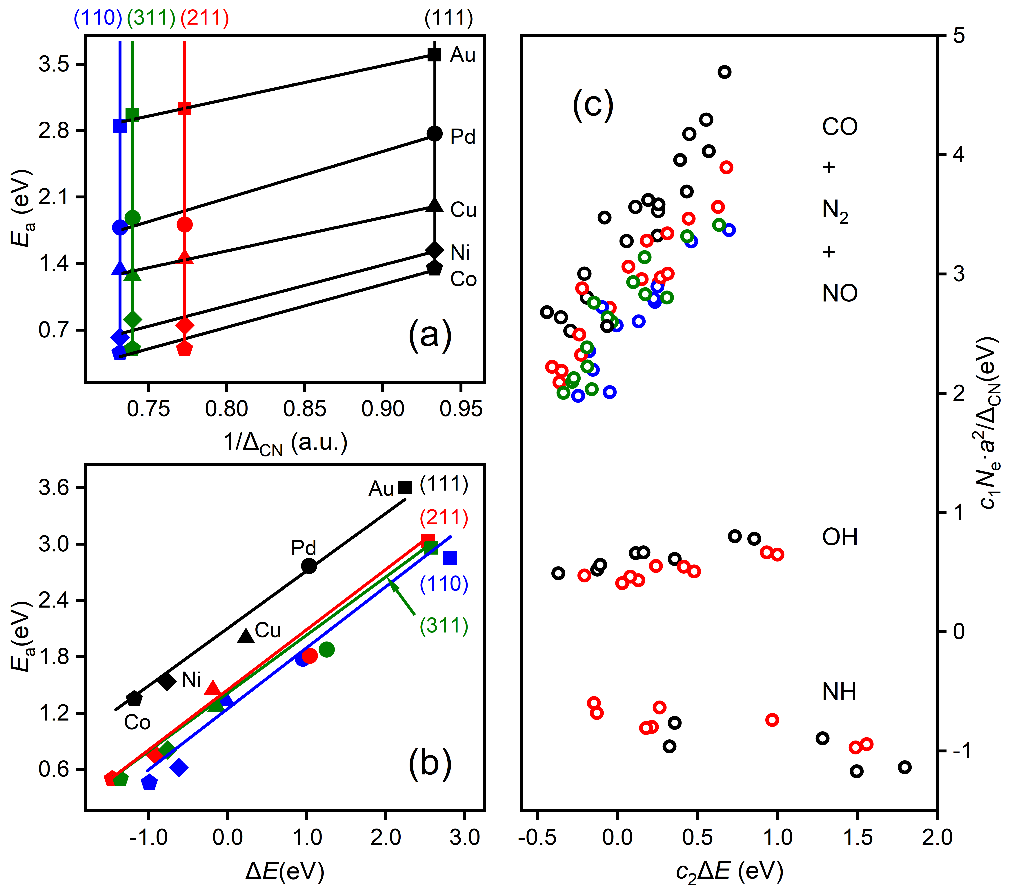
图3 活化能垒的几何效应与电子效应、结构效应与能量效应的去纠缠统计分析
中国科大博士生舒武为第一作者,中国科大的李微雪教授为通讯作者,上海大学的欧阳润海博士为共同通讯作者。该项工作得到了国家自然科学基金创新研究群体、面上项目、中国科学院先导项目、科技部项目资助,以及中国科学技术大学超算中心为该项目研究提供的机时。
文章链接:https://pubs.acs.org/doi/10.1021/jacs.4c01524
(中国科学技术大学化学与材料科学学院,精准智能化学重点实验室、合肥微尺度物质科学国家研究中心,科研部)

